
Daniela C. Ferrara, MD, MS, Sao Paulo, Brazil; K. Bailey Freund, MD, and Lawrence A. Yannuzzi, MD, New York City
Age-related maculopathy is a degenerative disorder of the macula, most often clinically apparent after 50 years of age, characterized by primary drusen deposition at the level of the retinal pigment epithelium. Age-related macular degeneration is defined as the late stage of ARM. While AMD is the leading cause of blindness and visual loss in the elderly in developed countries, the clinical and epidemiological complexity of the disorder presents a challenge in terms of creating a clinically meaningful classification scheme.1 There are currently different classifications for AMD, and the condition can be epidemiologically divided into two large subcategories: dry and wet AMD,2 which account for approximately 40 percent and 60 percent of the advanced cases respectively.1
The anatomic and functional damage in AMD is secondary to degenerative, atrophic and neovascular changes that affect the neurosensory retina and underlying choroid. Large confluent drusen and significant pigmentary changes within the retina are the major risk factors for both dry and wet AMD.1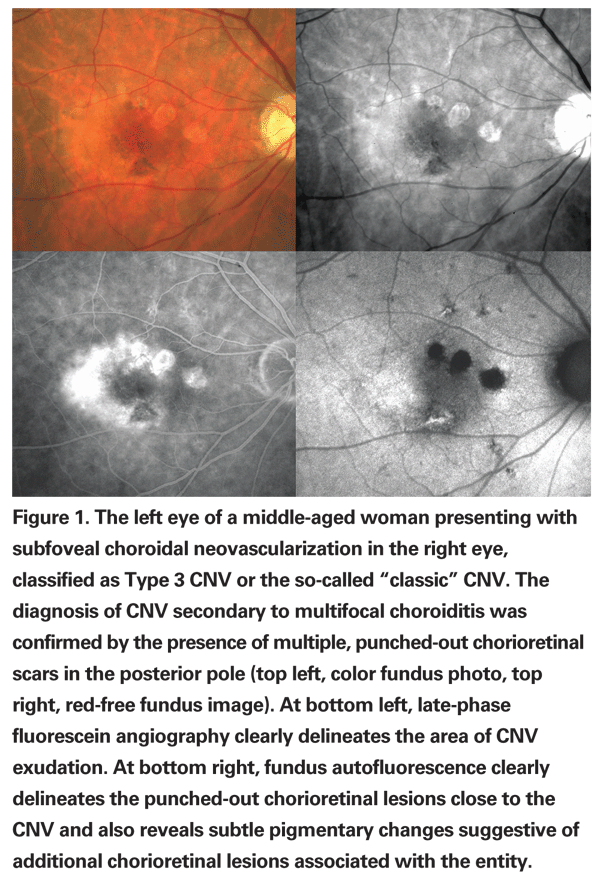
Wet, or exudative, AMD can be defined by the presence of retinal pigment epithelium detachment with or without associated neurosensory retinal detachment.2 Either subretinal or sub-RPE capillary proliferation and neovascular membrane formation constitute the neovascular form of exudative AMD. The diagnosis of wet AMD can also be based on the presence of epiretinal, intraretinal, subretinal, or sub-RPE scar tissue or fibrin-like deposits, which may eventually develop after resolved choroidal neovascularization.2 However, several degenerative and dystrophic diseases of the retina and RPE evolve with significant pigmentary changes and lipofuscin accumulation which may eventually be associated with RPE or neurosensory retinal detachment. Depending on the severity of the clinical presentation or on the stage, these conditions may mimic either wet or dry AMD. Therefore, it is necessary to screen for entities such as central serous chorioretinopathy, adult-onset vitelliform macular dystrophy (AVMD) and other pattern dystrophies of the RPE.
Angiogenesis and CNV are a final common pathway in numerous ophthalmic diseases as a stereotypic, nonspecific wound repair response triggered by variable factors. Secondary CNV may occur rarely in central serous chorioretinopathy, either spontaneously or as an iatrogenic event following laser treatment. The condition itself appears to be determined by a choroidal hyperpermiability state and characteristically causes a circumscribed pigmentary mobilization following the resolution of subretinal fluid. Although AVMD and other pattern dystrophies of the RPE also tend to evolve into pigmentary changes and RPE atrophy, neovascular complications may occasionally occur. Secondary CNV can also occur in different chorioretinal diseases, including chorioretinitis, pathologic myopia, trauma, angioid streaks and other hereditary macular dystrophies.
Novel diagnostic tools, as well as potential prophylactic and therapeutic targets, have been derived from an increasing understanding of pathophysiologic mechanisms in AMD. Early diagnosis and efficient intervention are crucial in the management of exudative AMD as well as in either inflammatory or dystrophic chorioretinal diseases, especially when complicated by CNV.
In last month's issue, we reviewed the conditions that mimic dry AMD. What follows is a review of the major diseases that mimic wet AMD to guide the comprehensive ophthalmologist in the differential diagnosis of these challenging cases.
Chorioretinitis and Idiopathic CNV
In a wide spectrum of inflammatory conditions named "white-dot syndromes," immune-mediated mechanisms can cause a variety of clinical presentations. Chorioretinal inflammatory lesions are the main clinical feature, and the precise diagnosis may sometimes be very difficult to differentiate from entities such as multifocal choroiditis, presumed ocular histoplasmosis syndrome and punctate inner choroidopathy. Moreover, as in idiopathic choroidal neovascularization, these conditions frequently affect young, otherwise healthy women with moderate or high myopia, and they share many other underlying immune-mediated features.3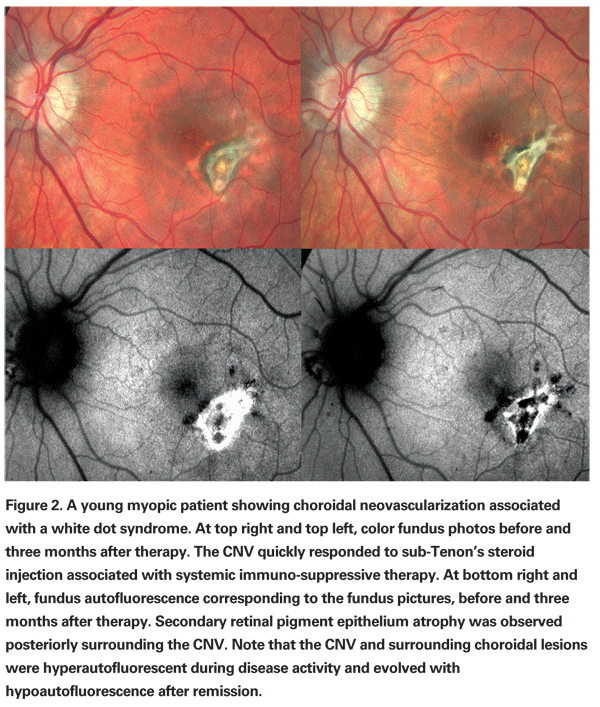
Multifocal choroiditis typically presents as a chronic relapsing panuveitis with multiple chorioretinal lesions. Acute yellow-white lesions primarily involve the choroid and outer retina, and usually evolve to punched-out chorioretinal scars with pigmented borders and peripapillary fibrosis.4 Clinical and angiographic manifestations of inflammation, such as vitreous cells and fluorescein staining of the optic nerve, help in making the correct diagnosis. Indocyanine green angiography and fundus autofluorescence photography may help identify areas of involvement around the optic nerve or in the periphery that may not be visible on clinical examination. Although frequencies vary in different series, CNV is the most common complication and the major cause of visual acuity loss in patients with multifocal choroiditis.4,5 (See Figures 1 and 2)
Angioid Streaks and PXE
Along with several hereditary dystrophic conditions associated with CNV, pseudoxanthoma elasticum has CNV as its major complication. Pseudoxanthoma elasticum (PXE) is a systemic disease of the connective tissue primarily affecting the cardiovascular system, the skin and the eye, caused by mutations in the ABCC6 gene and characterized by marked clinical heterogeneity. The underlying pathophysiologic mechanisms remain obscure.6 Characteristic skin changes most commonly affect the sides of the neck and axilla and help in suggesting the diagnosis, which is confirmed by skin biopsy.
Ophthalmologic manifestations of pseudoxanthoma elasticum are highly variable and include diffuse pigmentary mobilization of the RPE typically described as a peau d'orange fundus, pattern dystrophy of the macula and angioid streaks.7 Angioid streaks are a clinical manifestation of calcified ruptures within Bruch's membrane and are usually asymptomatic unless a secondary neovascular membrane evolves.6,8 Fundus autofluorescence imaging highlights the peau d'orange fundus, angioid streaks and the frequently associated optic nerve head drusen. Fundus autofluorescence imaging may also reveal RPE rips and large areas of missing or atrophic RPE associated with the streaks.9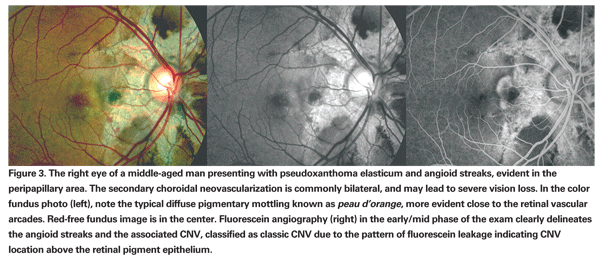
CNV can complicate angioid streaks in approximately 86 percent of the affected eyes (See Figure 3) and the streaks are usually bilateral. Fluorescein and indocyanine green angiography reveal the angioid streaks as well as an associated neovascular membrane. Secondary CNV in pseudoxanthoma elasticum has been associated with a poor visual prognosis.7,8
Pathologic Myopia
Pathologic myopia, or high myopia, is an important cause of secondary CNV in young people and a major cause of legal blindness in many developed countries (See Figure 4). The progressive elongation of the eye is associated with variable morphologic alterations within a posterior staphyloma. Macular CNV is the most common vision-threatening complication of high myopia, affecting 5 percent to 10 percent of patients. The long-term visual prognosis is very poor. However, distinct from AMD, the CNV in myopia tends to have less exudative activity and a self-limited course, resulting in smaller chorioretinal scars often referred to as a Fuchs' spot. Studies on the natural course of this entity reveal that the bleeding from CNV tends to absorb spontaneously in a few months, and recurrences are unusual. Typically, the myopic CNV regresses completely after a few years, becoming flat and sometimes unrecognizable. However, chorioretinal atrophy around the regressed scar is a frequent complication late in the natural course. It tends to gradually enlarge, contributing to progressive visual impairment over the disease evolution.10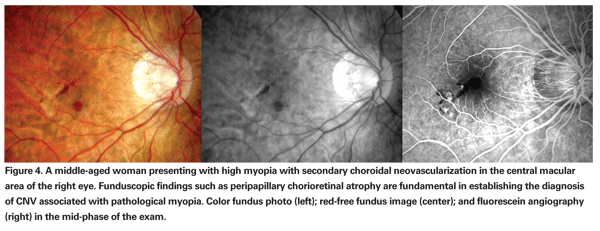
Other morphological features typical of pathologic myopia that help to distinguish it from AMD include: a myopic configuration of the optic nerve head with its associated myopic crescent; posterior staphyloma; vitreous degenerative changes; myopic or "tigroid" fundus degeneration; lacquer crack formation; choroidal atrophy; bare sclera; and lastly, peripheral retinal degeneration or retinal hole formation.
Idiopathic Macular Telangiectasia
Retinal capillary telangiectasia can develop in the macula without a known cause or can be secondary to retinal vasculitis, retinal vein occlusion or underlying systemic conditions such as diabetes, hypertension and infectious/inflammatory diseases.11
Donald M. Gass, MD, described a primary idiopathic form of these entities termed idiopathic juxtafoveal retinal telangiectasis (IJRT) and further proposed a classification with subgroups and stages.12,13 Lawrence A. Yannuzzi, MD, established the terminology idiopathic macular telangiectasia, essentially simplifying the Gass-Blodi model based on newly recognized clinical manifestations of the disease.11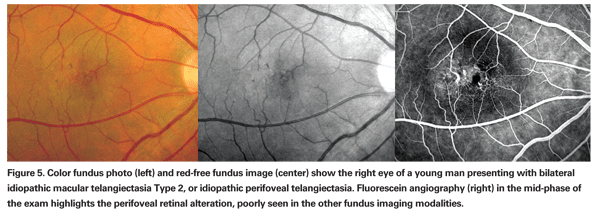
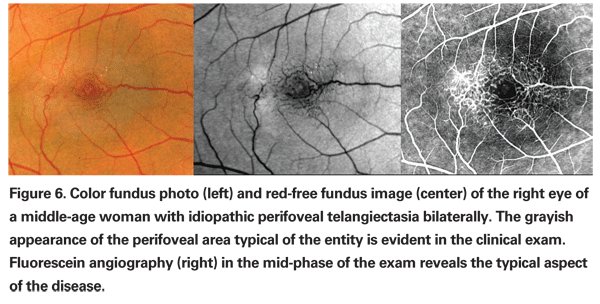
According to this new classification, Type 1, aneurysmal telangiectasia, is characterized by multiple capillary, venular and arteriolar aneurysms affecting the juxtafoveolar and the extramacular regions and also the peripheral fundus. It is typically unilateral and primarily affects males. Type 2 is defined by the presence of bilateral perifoveal telangiectasia; this manifestation of the disease is therefore named idiopathic perifoveal telangiectasia (IPT). Perifoveal telangiectasia has been subdivided into two stages: a nonproliferative stage with retinal exudation or atrophy (See Figures 5 and 6); and a proliferative stage in which subretinal neovascularization occurs (See Figure 7). Type 3, an occlusive telangiectasia, is very rarely seen, and it is still disputable whether it is a true variant.11-13
Vision loss in Type 1 occurs secondary to exudative changes such as intraretinal edema and cystic changes, subretinal exudation and lipid deposition in the fovea. While these exudative findings may mimic wet AMD, the presence of retinal aneurysms, earlier age of onset, absence of AMD features in the fellow eye and lack of angiographic evidence of CNV are distinguishing features.
In the nonproliferative stage of IPT, intraretinal leakage or staining evident on fluorescein angiography may mimic wet AMD. Clinical features such as "right-angle" venules, superficial retinal crystalline deposits, intraretinal pigment migration and perifoveolar loss of retinal transparency are distinguishing features. Angiographic evidence of temporal juxtafoveolar capillary telangiectasia in the early phase study and absence of CME or subretinal fluid on OCT are also helpful in identifying this stage of IPT. Identification of the proliferative stage of IPT is even more challenging, particularly in patients over age 50. When the proliferative changes are unilateral, recognition of the nonproliferative findings in the fellow eye can be help in avoiding misdiagnosis.11
Idiopathic PCV
Idiopathic polypoidal choroidal vasculopathy, or simply polypoidal choroidal vasculopathy (PCV), is a peculiar form of CNV with distinct features that distinguish it from typical wet AMD. Among them are different risk factors, clinical features, angiographic findings, natural course and visual prognosis.11,14,15 The actual prevalence of PCV is still unknown, given the tremendous difficulty in differentiating the diagnosis from typical AMD on a clinical basis alone.
PCV was originally described as an exudative form of macular degeneration.14 The characteristic clinical presentation has multiple serous and hemorrhagic detachments of the neurosensory retina and RPE and is associated with a distinct network of branching abnormal vessels beneath the choriocapillaris with terminal aneurysmal dilations. These terminal polyps are occasionally evident on clinical examination as a reddish-orange, spheroid, polyp-like structure. If there is some extent of RPE atrophy above the lesions, the polyps may also be seen on fluorescein angiography, although indocyanine-green angiography is the diagnostic method of choice to image the lesions and rule out AMD or CSC (See Figure 8).14,16 Polypoidal lesions are typically located bilaterally in the peripapillary area but may occur in the macular area, in the periphery or may be widespread in the fundus. Drusen are less frequently seen in PCV eyes than in more typical AMD cases.16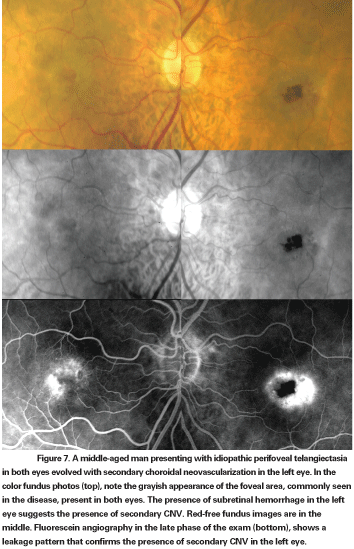
The natural course may be associated with persistent or recurrent serosanguineous detachments of the neurosensory retina and RPE but some polyps evident on angiogram may remain stable and asymptomatic. Patients with PCV can retain good vision in the long-term, even when it affects the macula.14 A vision-threatening complication is the occurrence of massive subretinal bleeding; when present, this suggests the diagnosis of PCV rather than typical neovascular AMD.16 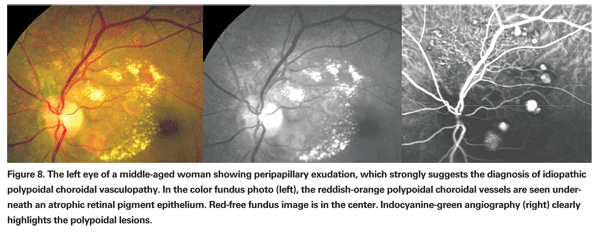
Novel therapeutic strategies such as intravitreal anti-VEGF therapy have been shown to significantly improve vision in conditions complicated by CNV, especially when performed early in the course of the disease. Distinguishing the wet form of AMD from the diseases that mimic it may have significant implications for the visual outcome. Since many of these conditions have either associated systemic features or demand associated systemic therapy, correct diagnosis may benefit a patient's general health.
Supported by The Macula Foundation Inc. The authors have no financial interest in this report.
Dr. Ferrara did a retina fellowship at the LuEsther T. Mertz Retinal Research Center, New York City, and is a now doctoral candidate in the Department of Ophthalmology, Otolaryngology and Head and Neck Surgery at the University of Sao Paulo. Drs. Freund and Yannuzzi practice at Vitreous-Retina-Macula Consultants of New York, and Dr. Yannuzi is in the Department of Ophthalmology at Columbia University. Contact Dr. Freund at 460 Park Ave. 5th floor, New York, NY 10022. Phone: (212) 861-9797, fax: (212) 628 0698, e-mail: mail@ vrmny.com.
1. Klein ML, Ferris III FL, Armstrong J, et al. AREDS Research Group. Retinal precursors and the development of geographic atrophy in age-related macular degeneration. Ophthalmology 2008;115:1026-1031.
2. Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. Surv Ophthalmol 1995;39:368-374.
3. Machida S, Fujiwara T, Murai K, Kubo M, Kurosaka D. Idiopathic choroidal neovascularization as an early manifestation of inflammatory chorioretinal diseases. Retina 2008;28:703-710.
4. Thorne JE, Wittenberg S, Jabs DA, et al. Multifocal choroiditis with panuveitis – Incidence of ocular complications and loss of visual acuity. Ophthalmology 2006;113:2310-2316.
5. Vianna RN, Ozdal PC, Filho JP, et al. Longterm follow-up of patients with multifocal choroiditis and panuveitis. Acta Ophthalmol Scand 2004;82:748-753.
6. Chassaing N, Martin L, Calvas P, Le Bert M, Hovnanian A. Pseudoxanthoma elasticum: a clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J Med Genet 2005;42:881-892.
7. Gass JDM. Stereoscopic atlas of macular diseases: diagnosis and treatment. 4th ed. St. Louis: Mosby, 1997:52-70.
8. Bhatnagar P, Freund KB, Spaide RF, et al. Intravitreal bevacizumab for the management of choroidal neovascularization in pseudoxanthoma elasticum. Retina 2007;27:897-902.
9. Sawa M, Ober MD, Freund KB, Spaide RF. Fundus autofluorescence in patients with pseudoxanthoma elasticum. Ophthalmology 2006;113:814-820.
10. Yoshida T, Ohno-Matsui K, Yasuzumi K, et al. Myopic choroidal neovascularization: a 10-year follow-up. Ophthalmology 2003;110:1297-1305.
11. Yannuzzi LA, Bardal AMC, Freund KB, Chen KJ, Eandi CM, Blodi B. Idiopathic Macular Telangiectasia. Arch Ophthalmol 2006;124:450-460.
12. Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol 1982;100:769-780.
13. Gass JD, Blodi BA. Idiopathic juxtafoveolar retinal telangiectasis: update of classification and follow-up study. Ophthalmol 1993;100:1536-1546.
14. Yannuzzi LA, Sorenson J, Spaide RF, Lipson B. Idiopathic polypoidal choroidal vasculopathy (IPCV). Retina 1990;10:1-8.
15. Yannuzzi LA, Ciardella A, Spaide R, Rabb M, Freund KB, Orlock D. The expanding clinical spectrum of idiopathic polypoidal choroidal vasculopathy. Arch Ophthalmol 1997;115:478-485.
16. Yannuzzi LA, Wong DW, Sforzolini BS, et al. Polypoidal choroidal vasculopathy and neovascularized age-related macular degeneration. Arch Ophthalmol 1999;117:1503-1510.







