
US Pharm.
2006;11:HS-17-HS-28.
Seizures
are common during childhood.1-9 Each year, approximately 150,000
pediatric patients experience their first seizure, and 30,000 are found to
have epilepsy. In addition, an estimated 4% to 10% of children have recurrent
unprovoked seizures by age 18.1
SEIZURES VERSUS EPILEPSY
Seizures are
paroxysmal, time-limited changes in motor activity and/or behavior that result
from abnormal electrical activity in the brain.1,3,10 Seizures can
either be localized (affecting one part of the brain) or widespread (affecting
the entire brain).1,3 Most seizures in children are provoked by
somatic disorders originating outside the brain (e.g., high fever, infection,
syncope, head trauma, hypoxia, toxins, cardiac arrhythmias).3,5 The
clinical appearance of a seizure depends on the location and extent of brain
involvement. For example, a seizure originating in the motor cortex may cause
only focal facial twitching; whereas a seizure involving the entire brain will
manifest as a generalized seizure with loss of consciousness.11
A child's age can serve as a clue to the
possible cause of the seizure.6,7 Children from infancy through
early school-age usually develop seizures due to fever, central nervous system
(CNS) infections, electrolyte and metabolic abnormalities, toxic ingestions,
and CNS tumors. In contrast, adolescents and teenagers experience first-time
seizures due to toxic ingestions, illicit drug or alcohol use, brain tumors,
or excessive video game use.7
Epilepsy is defined as two or
more unprovoked seizures occurring at an interval greater than 24 hours apart.3,8
Less than one third of seizures in children are categorized as epileptic.3
Provoked seizures, such as those resulting from acute head trauma or
meningitis, do not constitute epilepsy.11
CLASSIFYING SEIZURES
Classification of
seizure disorders has evolved considerably through the years. The terms grand
mal or petit mal have been replaced by more detailed
classifications based on specific clinical manifestations, extent of brain
involvement (i.e., focal or generalized), and discrete clinical syndromes.6
Childhood epilepsy is usually
classified using variations of the International Classification of Epileptic
Seizures. This nomenclature delineates seizures into two categories:
generalized and partial (focal) seizures.9,11-13
Generalized Seizures
Generalized
seizures originate in a bilaterally symmetric fashion within the brain and can
be further classified as convulsive or nonconvulsive seizures.
Convulsive seizures include
tonic, clonic, and tonic-clonic seizures. Tonic-clonic seizures are
characterized by an abrupt loss of consciousness with tonic extension of the
trunk and extremities (tonic phase), followed by synchronous muscle jerking
(clonic phase). In some patients, only a tonic or clonic phase is apparent.
During the postictal period, patients are confused and sleepy and may complain
of headache. Although generalized seizures are not preceded by an aura (e.g.,
noxious smell or taste, unusual epigastric sensation), many patients may
experience nonspecific premonitory symptoms (e.g., dizziness, irritability,
anxiety).
Nonconvulsive seizures include
absence, myoclonic, and atonic seizures. Absence seizures are characterized by
brief attacks--usually lasting five to 10 seconds--of arrest of consciousness
and movement. Some minor movements such as lip smacking or blinking may be
observed. Absence seizures are not associated with postictal drowsiness.
Episodes that are prolonged or have more prominent movements are termed atypical
absence seizures. Myoclonic seizures are characterized by brief, single or
repetitive muscle contractions of a muscle or group of muscles. Atonic
seizures are characterized by a sudden, momentary loss of muscle tone or
posture.11
Partial Seizures
Partial seizures originate in one
part of the brain and do not produce a complete loss of consciousness.11
However, they may subsequently progress to a generalized seizure.3,11,12
Partial seizures can be further subdivided into simple and complex partial
seizures. Simple partial seizures do not produce alternations of
consciousness, whereas complex partial seizures do.3,11 Auras
(i.e., complex partial or secondarily generalized seizures) are simple partial
seizures that precede other seizure types.11
CHILDHOOD EPILEPSY SEIZURES
The vast majority
of epilepsy syndromes begin during infancy and childhood.2
Determining which epileptic syndrome a child has is essential in establishing
prognosis and providing treatment.11 Common epileptic syndromes of
infancy and childhood include febrile seizures, neonatal seizures, infantile
spasms, Lennox-Gastaut syndrome, and childhood and juvenile absence epilepsy.
Febrile Seizures
Febrile seizures
are the most common childhood seizure disorder and generally have an excellent
prognosis; however, they may signify a serious underlying acute infectious
disease such as sepsis or bacterial meningitis.3 Febrile seizures
occur in approximately 3% of children ages 6 months to 6 years (peak age, 18
to 24 months). Most febrile seizures are "simple"--i.e., single, brief (less
than 15 minutes in duration), and generalized. Approximately one third of
febrile seizures are "complex" (multiple occurrences within 24 hours,
prolonged, or focal).1 Some children who experience simple febrile
seizures--including those who have an initial febrile seizure before age 9
months, are developmentally delayed, have a family history of afebrile
seizures, or have a preexisting neurological disorder--may be at risk for
epilepsy.1,3
Routine treatment of febrile
seizures involves searching for the cause of the fever and taking measures to
control the fever (e.g., with the use of antipyretics). Most children with
febrile seizures do not require anticonvulsant drugs.1 In fact,
anticonvulsant prophylaxis for preventing recurrent febrile convulsions is not
recommended.3
Neonatal Seizures
Neonates are at particular risk for
seizures because metabolic, structural, and infectious diseases are more
likely to manifest at this age than at any other.3 Neonates develop
seizures primarily because of CNS infections, electrolyte abnormalities (e.g.,
hypocalcemia, hypoglycemia), hypoxic ischemic encephalopathy, and rarely,
pyridoxine deficiency. These seizures can sometimes be a symptom of a more
serious condition (e.g., birth trauma, congenital structural brain
abnormalities, inborn errors of metabolism).
Unfortunately, neonatal
seizures are associated with a high rate of morbidity and mortality and can be
difficult to recognize. An infant experiencing neonatal seizures may display
only subtle changes (e.g., apnea, sustained eye deviation, chewing, or limb
bicycling movements). Overall, the rate of mental retardation and cerebral
palsy in survivors of neonatal seizures is between 15% and 45%. Prognosis and
treatment options are dependent on the etiology of the seizure.11
Infantile Spasms (West
Syndrome)
Infantile spasms
present during the first year of life and consist of rapid, jackknife flexor
or extensor spasms that appear in clusters. Because children with infantile
spasms tend to cry and draw up their legs during an attack, an initial
diagnosis of colic is common. Approximately 67% of children who have infantile
spasms have an underlying CNS disorder (e.g., congenital brain malformation,
tuberous sclerosis). The outcome of infantile spasms is usually poor--only
about 50% of children with infantile spasms attain remission, while 90% to 95%
become mentally retarded. The prognosis is substantially improved if
development is normal before onset of the spasms. First-line treatment for
infantile spasms consists of adrenocorticotropic hormone or prednisone.11
Lennox-Gastaut Syndrome
Lennox-Gastaut
syndrome is a severe form of epilepsy that is characterized by mental
retardation, multiple seizure types, and a classic electroencephalographic
(EEG) pattern of slow spike and wave.9,11 While mental retardation
may not be present at the onset of seizures, it eventually develops in 78% to
96% of patients. The age at onset of Lennox-Gastaut syndrome is between 1 and
6 years. This epileptic syndrome may evolve from infantile spasms--30% of
patients with infantile spasms develop Lennox-Gastaut syndrome, and 20% of
patients with Lennox-Gastaut syndrome have a history of infantile spasms.10
Patients with Lennox-Gastaut syndrome usually experience frequent seizures of
multiple types, including atonic, atypical absence, myoclonic, tonic-clonic,
and partial seizures.
First-line treatment for
Lennox-Gastaut syndrome consists of valproic acid; however, this drug alone
rarely controls the condition.11 Other treatments for
Lennox-Gastaut syndrome include lamotrigine, topiramate, felbamate, and the
ketogenic diet. Felbamate was the first antiepileptic medication approved for
the treatment of Lennox-Gastaut syndrome. Because felbamate can cause liver
failure and aplastic anemia, it should be reserved for patients who do not
adequately respond to alternative agents and whose condition is so severe that
the benefits outweigh the risks.14-17 Overall, the prognosis in
patients with Lennox-Gastaut syndrome is poor.11
Childhood and Juvenile
Absence Epilepsy
Childhood and
juvenile absence epilepsy begins most commonly between ages 4 and 12 years and
is characterized by frequent recurrent absence seizures. Generalized
convulsive seizures occur in up to 50% of patients. Treatment for childhood
and juvenile absence epilepsy consists of ethosuximide or valproic acid
(valproic acid is used in patients who have generalized tonic-clonic
seizures). The prognosis for this condition is excellent--children usually
outgrow this pattern and development is normal. Hyperventilation can trigger
absence spells and can be used to diagnose absence epilepsy.11
Status Epilepticus
Status epilepticus
is characterized by more than 30 minutes of unconsciousness and continuous or
intermittent seizure activity and is considered to be a true neurologic
emergency.11,13 The condition occurs more frequently in
children--especially those younger than 1 year--and is associated with high
morbidity and mortality. Among children, the most common precipitant of status
epilepticus is febrile illness, followed by medication change, idiopathic
epilepsy, metabolic derangements, and congenital abnormalities.11
The first step in the
management of status epilepticus is to support vital functions. The airway
should be protected, and the patient's vital signs (i.e., through continuous
oximetry and electrocardiography) should be closely monitored. Supplemental
oxygen at a rate of approximately 4 L per minute is recommended. Intravenous
(IV) access should be secured for the administration of parenteral
medications, and blood should be drawn for a complete blood count and to
measure electrolyte, glucose, calcium, magnesium, and anticonvulsant drug
concentrations. A toxicology screen should be performed as well. Medications
that can potentiate or precipitate status epilepticus are listed in Table 1
.3,13

Parenterally administered
benzodiazepines, particularly diazepam and lorazepam, are usually the initial
drugs used in the treatment of status epilepticus. Seventy-five percent to 90%
of the time, these agents diffuse quickly into the CNS and rapidly terminate
seizure activity. Both diazepam and lorazepam are equally effective in
terminating status epilepticus, but lorazepam has a substantially longer
duration of antiseizure effect (12 to 24 hours vs. 15 to 30 minutes). The
recommended pediatric IV dose of lorazepam is 0.1 mg/kg (maximum dose, 8 mg)
at a rate of 1 to 2 mg per minute. The recommended IV dose of diazepam is 0.2
to 0.5 mg/kg (maximum dose, 10 mg) at a rate of 2 mg per minute. If IV access
cannot be obtained, diazepam can also be administered rectally at a dose of
0.5 mg/kg (maximum dose, 20 mg).
Simultaneous loading with
phenytoin or fosphenytoin is also recommended. If seizures continue despite
these measures, phenobarbital should be administered parenterally. As a last
resort, barbiturate coma, high-dose midazolam, high-dose propofol, or general
anesthesia with neuromuscular blockade should be employed.13
TREATING SEIZURES IN CHILDREN
Determining whether seizure-like
symptoms in children constitute a true seizure disorder is important in the
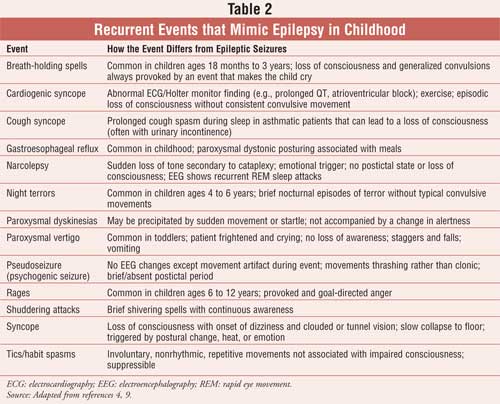
management of childhood epilepsy syndromes (see Table 2). Thirty
percent of children who have a seizure have a second seizure, and those
children who have electroencephalogram (EEG) abnormalities, previous
neurologic injury, partial seizures, and/or a family history of seizures are
more likely to have additional seizures.3,13
Once a seizure disorder has been accurately
diagnosed, treatment should be started as soon as possible. Both pharmacologic
and nonpharmacologic therapies have shown efficacy in treating seizures in
children.
Pharmacologic Therapy
Choosing the best
medication for a pediatric patient with a seizure depends upon many factors,
including patient age, seizure type, general health, concurrent disease states
and medication use, and cost.7 Clinicians commonly refrain from
prescribing antiepileptic medications to previously healthy children with a
first afebrile seizure who are not at risk for a second seizure.4
Most pediatric seizures are
controlled with the first antiepileptic drug that is selected. In order to
increase compliance and decrease side effects, antiepileptic drugs should be
started at the lowest possible dose and increased slowly. If satisfactory
control is not established within the first three to six months, a second
antiepileptic drug should be added, with the eventual goal of eliminating the
first agent and achieving monotherapy.1
The antiepileptic medications
can be divided into two categories: first- and second-generation drugs. The
following section describes the mechanism of action, place in therapy,
pediatric dose, and therapeutic range of the first- and second-generation
antiepileptic drugs. Side effects and drug interactions associated with these
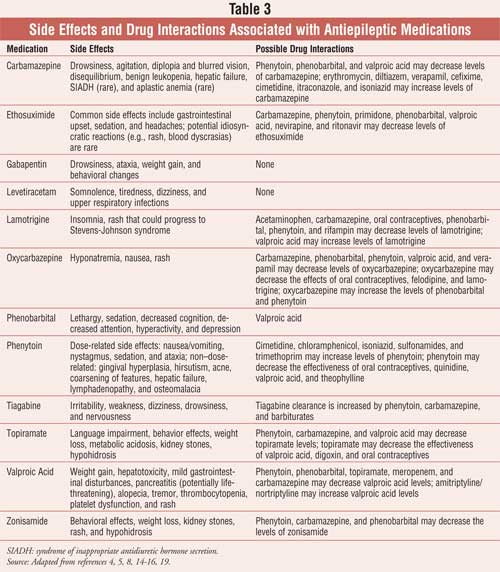
products are listed in Table 3.
First-Generation
Antiepileptic Drugs: Phenytoin, phenobarbital, carbamazepine,
ethosuximide, and valproic acid are considered first-generation antiepileptic
drugs. These products have well-established therapeutic ranges of blood levels
but are associated with manyside effects and drug interactions.
Phenytoin--which acts by
decreasing the sustained repetitive firing of single neurons by blocking
sodium-dependent channels and decreasing depolarization-dependent calcium
uptake--is used for primary and secondary generalized tonic-clonic seizures,
partial seizures, and status epilepticus.4 Pediatric maintenance
doses of phenytoin average 5 to 10 mg/kg/day, and a therapeutic range of blood
levels of 10 to 20 mg/dL is usually desired. Fosphenytoin, the water-soluble
prodrug of phenytoin, is generally preferred over IV phenytoin when immediate
loading doses are needed (e.g., status epilepticus).3,4 Fosphenytoin
can be more safely administered via peripheral IV than can phenytoin.4
Phenobarbital and primidone,
which is metabolized to phenobarbital, act on the gamma-aminobutyric acid
(GABA) receptor to increase the chloride channel open duration.3,14
Phenobarbital is particularly useful for generalized tonic-clonic seizures in
children and is dosed at 5 to 10 mg/kg/day. The therapeutic range of
phenobarbital is considered to be 15 to 40 mg/dL.3,4
Carbamazepine, which has a
mechanism of action similar to that of phenytoin, is effective for the
management of generalized tonic-clonic and partial seizures.3 The
drug is usually given orally in doses of 10 to 20 mg/kg/day, and a therapeutic
range of blood levels of 8 to 12 mg/dL is commonly desired. Drug metabolism
typically increases after the first month of therapy, owing to hepatic
autoinduction.4
Ethosuximide provides its
anticonvulsant action by blocking calcium channels associated with
thalamocortical circuitry. This product is considered an effective drug for
the management of typical absence epilepsy.3 The typical target
pediatric dose is 10 to 20 mg/kg/day, and therapeutic blood levels in the
range of 40 to 100 mg/dL are usually desired.4
Valproic acid is defined as a
broad-spectrum anticonvulsant. It acts by blocking voltage-dependent sodium
channels and increasing calcium-dependent potassium conductance. This drug is
useful for the management of many seizure types, including generalized
tonic-clonic, absence, atypical absence, and myoclonic seizures.3
Valproic acid is available orally and intravenously and is initially dosed at
10 to 20 mg/kg/day; however, doses of 100 mg/kg/day have been documented.14
When measured, therapeutic blood levels of valproic acid should be in the
range of 50 to 100 mg/dL.4
Second-Generation
Antiepileptic Drugs: Gabapentin, lamotrigine, levetiracetam,
oxcarbazepine, tiagabine, topiramate, and zonisamide constitute the
second-generation antiepileptic drugs. Because these products have very little
effect on the cytochrome P-450 enzyme system, they are not associated with as
many drug interactions. However, the second-generation products lack
established therapeutic ranges of blood levels and are considerably more
expensive than the first-generation antiepileptic products.7,15
Gabapentin exerts its
antiepileptic action by binding to neuronal membranes (glutamate synapses) and
increasing brain GABA turnover.3 This product is used as an add-on
drug for patients with refractory complex partial and secondary generalized
tonic-clonic seizures.3,4,7 Gabapentin is recommended for children
12 years and older.3 The typical target dose of gabapentin is 20 to
40 mg/kg/day, and drug concentrations are not routinely measured.4
Lamotrigine affects
voltage-sensitive sodium channels and inhibits the presynaptic release of
glutamate and aspartate.3,14 It is primarily used as an add-on drug
for the management of complex partial and generalized tonic-clonic seizures in
children older than 2 years.3,7 Doses of lamotrigine range from 5
to 15 mg/kg/day.4 Lamotrigine does not require laboratory testing.7
Levetiracetam can be used to
treat partial onset seizures in children older than 4 years. Doses of
levetiracetam begin at 20 mg/kg/day. This drug does not require blood level
monitoring.14,18 The mechanism of action of levetiracetam is
unknown.3
Oxcarbazepine, which has a
mechanism of action similar to those of phenytoin and carbamazepine, is useful
as adjunctive therapy for children with partial seizures.3 This
product is usually dosed in the range of 10 to 30 mg/kg/day and does not
routinely require laboratory monitoring.4 Blood levels are
infrequently measured in clinical practice.
Tiagabine inhibits seizure
activity by blocking reuptake of the neuroinhibitory transmitter GABA into
neuronal and glial cells.3 This drug is effective in the management
of complex partial seizures as an add-on drug in children older than 12 years.3,7
It is most commonly dosed at 1 to 2 mg/kg/day and does not require laboratory
testing.4
Topiramate produces
anticonvulsant action by blocking voltage-dependent sodium channels. This drug
is used as adjunctive therapy for refractory complex seizures with or without
secondary generalization.3 Topiramate is given to children older
than 2 years in doses of 1 to 9 mg/kg/day.4,7 Blood levels are
infrequently measured in clinical practice.
Zonisamide is useful as an
adjunctive treatment for partial seizures and may also be useful for myoclonic
syndromes in children older than 16 years.3 A dosage of 5 to 10
mg/kg/day is commonly given to children, and blood level monitoring is not
required.4 The mechanism of action of zonisamide is unclear.
Pediatric Dosing
Considerations: Throughout the neonatal period, drug absorption and
clearance may change, requiring close monitoring of levels and adjustment of
antiepileptic drug dosages. These alterations in pharmacokinetics--as well as
changes in metabolic rates, hepatic and renal function, and body
mass--continue throughout childhood. More rapid clearance and variability in
elimination kinetics of antiepileptic drugs can also affect therapy--children
often require higher dosages than those recommended for adults. More frequent
dosing may also be needed to avoid low therapeutic blood levels throughout the
day. In refractory patients, dosage and therapeutic blood levels should be
followed closely. In late childhood and adolescence, therapeutic blood levels
tend to remain fairly constant, although adjustments may be required around
the time of puberty and during growth spurts.
Nonpharmacologic Therapy
The ketogenic
diet--a high-fat, low-carbohydrate, adequate-protein diet--has gained
popularity in recent years as a method of managing seizures.15-17
It has been shown to reduce seizure frequency and to decrease the
antiepileptic drug burden and is used primarily to treat children with
symptomatic types of epilepsy (e.g., infantile spasms, Lennox-Gastaut
syndrome, progressive myoclonic epilepsy). Some data suggest that it may also
be effective in the treatment of intractable partial and generalized epilepsy.
However, about 10% of patients who practice the ketogenic diet experience side
effects, including hypoproteinemia, hemolytic anemia, and kidney stones.15
Much remains unknown about the ketogenic diet, including its mechanism
of action, the optimal protocol, and its full range of applicability.15
Surgical procedures (e.g.,
resective surgery, vagus nerve stimulation) are another form of
nonpharmacologic therapy used to treat children with seizures. These
procedures can be used in pediatric patients who suffer from ongoing seizures
or who cannot tolerate antiepileptic drugs; however, because of their
associated risks (e.g., stoke, sudden death, infection, vocal cord paralysis),
they are used as a last resort.15,17
REFERENCES
1. McAbee GN, Wark JE. A practical approach to uncomplicated seizures in children. Am Fam Physician. 2000;62:1109-1116.
2. Hirsch E. Childhood epilepsy syndromes with both focal and generalized seizures. Acta Neurol Scand Suppl . 2005;181:52-56.
3. Behrman RE, Kliegman RM, Jenson HB. Nelson Textbook of Pediatrics. 17th ed. Phildelphia, PA: Elsevier Science; 2004.
4. Robertson J, Shilkofski N. The Harriet Lane Handbook. 17th ed. Philadelphia, PA: Elsevier; 2005.
5. Wallace SJ, Farell K. Epilepsy in Children. 2nd ed. London: Arnold; 2004.
6. Hoban TF. Seizure Disorders in Childhood. Loyola University Chicago Stritch School of Medicine Web site. Available at: www.meddean.luc.edu/lumen/MedEd/pedneuro/epilepsy.htm. Accessed March 2006.
7. Flomin O, Nield L, Kamat D. Seizure medications: a review for the primary care pediatrician. Clin Pediatr (Phila). 2005;44:383-391.
8. Spencer DC, Roberts CM. Stopping seizure medications in children: when is it safe? Neurology. 2005;64:E32-E33.
9. Murphy JV, Dehkharghani F. Diagnosis of childhood seizure disorders. Epilepsia. 1994;35 Suppl 2:S7-S17.
10. Pugh MB, Werner B, Filardo TW, et al. Stedman's Medical Dictionary. 27th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000.
11. Marx JA, Hockberger RS, Walls RM. Rosen's Emergency Medicine: Concepts and Clinical Practice. 5th ed. St Louis, MO: Mosby, Inc; 2002.
12. Chang BS, Lowenstein DH. Epilepsy. N Engl J Med. 2003;349:1257-1266.
13. Rakel RE. Textbook of Family Practice. 6th ed. Phildelphia, PA: W.B. Saunders Company; 2002.
14. Armstrong LL, Goldman MP, Lance L. Drug Information Handbook. 10th ed. Hudson, OH: Lexi Comp, Inc.; 2002.
15. Nadkarni S, LaJoie J, Devinsky O. Current treatments of epilepsy. Neurology. 2005;64:S2-S11.
16. Sinha SR, Kossoff EH. The ketogenic diet. Neurologist. 2005;11:161-170.
17. Faught RE. Management of pediatric epilepsy. Highlights of the American Epilepsy Society 58th Annual Meeting 2005. Available at: www.medscape.com/viewarticle/496930.
18. Shields WD, Koh S. The Role of
Newer Antiepileptic Drugs in Children with Epilepsy. Medscape 2000. Available
at: www.medscape.com/viewprogram/309?src=search.
To comment on this article, contact
[email protected].







