
 A 42-year-old black female presented for an eye examination with a chief complaint of painless, intermittent red eyes that had persisted for two months. Her systemic history was significant for Crohn’s disease and sarcoidosis. There was no history of any ocular disease or trauma.
A 42-year-old black female presented for an eye examination with a chief complaint of painless, intermittent red eyes that had persisted for two months. Her systemic history was significant for Crohn’s disease and sarcoidosis. There was no history of any ocular disease or trauma.
Diagnostic Data
Her best-corrected visual acuity was 20/25 O.U. at distance and near. External examination was normal, with no evidence of afferent pupillary defect.
Her intraocular pressure measured 14mm Hg O.U. Dilated fundoscopy was within normal limits in both eyes. There was no evidence of vasculitis or pars planitis; however, we noted the presence of vitreous cells (most likely secondary to anterior cell “spillover”). The pertinent anterior segment findings are illustrated in the photographs.
Your Diagnosis
How would you approach this case? Does this patient require any additional tests? What is your diagnosis? How would you manage this patient? What’s the likely prognosis?
Discussion
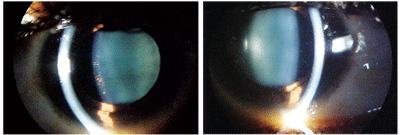
This patient with a history of
Crohn’s disease and sarcoidosis presented with a chief complaint of
painless, intermittent red eyes (O.D left, O.S. right).
Additional testing might include checking the mouth for ulcers and obtaining additional history regarding the genitourinary and digestive tracts to rule out Behçet's disease. Additional procedures might include gonioscopy to rule out angle obstruction secondary to debris or peripheral anterior synechiae formation.
The diagnosis in this case is granulomatous panuveitis, presumably secondary to sarcoidosis. Sarcoidosis is one of the leading causes of uveitis in North America—20% to 40% of patients with sarcoidosis develop uveitis.1 Anterior uveitis is a common finding in patients who are diagnosed with sarcoidosis and can serve as a significant first clue to the discovery of undiagnosed cases. Anterior inflammation is more common in patients with darker pigmentation, while white patients with sarcoidosis tend to develop posterior uveitis or panuveitis.1
Sarcoidosis is a systemic disorder characterized by localized granulomatous formations and various clinical features, depending upon geographic regions and ethnic groups. Ocular involvement is common and may precede clinical manifestations of systemic disease. Topical and oral corticosteroids are the mainstay of treatment for ocular and systemic manifestations of sarcoidosis, and they can effectively prevent severe visual impairment.1,2 Other oral immunomodulatory agents can be added as necessary or serve as alternatives to steroid intolerant patients.
Following traumatic iridocyclitis, the most common causes of anterior uveitis are idiopathic, the seronegative spondyloarthropathies, juvenile rheumatoid arthritis and herpetic keratouveitis. The vast majority of intermediate uveitis cases are secondary to idiopathic, tuberculosis, syphilis, multiple sclerosis, lyme disease and large reticulum cell sarcoma.3 When testing is negative and no associated underlying condition can be found, by exclusion, the cause may be labeled as idiopathic.3,4 A comprehensive history and testing will help insure a proper diagnosis for all patients who present with uveitis.
Clinical manifestations of uveitis vary depending on the primary site of involvement, the course of the inflammatory process and the presence of secondary complications arising from the uveitis itself.
Symptoms of acute anterior uveitis include pain, diffuse redness, photophobia and blurred vision that may develop over a period of hours or days. Chronic uveitis may present with simple blurred vision and/or mild redness with little pain. Patients who have posterior uveitis may present with floaters or impaired vision, particularly if the posterior pole is involved. Patients who have panuveitis may present with any or all of these symptoms.
Clinical examination findings also vary depending on the location, course and pathogenesis of the inflammation. Virtually all ocular tissues have the potential to be affected and yield findings that could be helpful in diagnosing the patient. The conjunctiva may show ciliary flush or nodules. Examine the cornea for keratic precipitates (small keratic precipitates are usually seen in nongranulomatous types of uveitis, while larger mutton-fat keratic precipitates are characteristic of granulomatous uveitis). New keratic precipitates are usually white, while old keratic precipitates become more pigmented or shrunken as they age.3
The mechanisms of inflammation occur at the cellular level (release of chemotactic factors and mediators that increase vascular permeability) and result in the presence of cells and flare in the aqueous humor. Both of these elements are graded on a scale of zero to four, with zero denoting the lowest level of severity. In severe cases of anterior uveitis, a fibrin clot of cells and chemoatractants may cause a dense settling of debris in the anterior chamber (hypopyon).1-3
The iris may show either anterior or posterior synechiae. If advanced, pupillary block, iris bombé and/or angle-closure glaucoma may result. Iris nodules (Koeppe nodules at the pupillary border and Busacca nodules within the iris stroma) or actual granulomas may be seen in cases of granulomatous uveitis.1,3 Additionally, cataract formation—particularly posterior subcapsular cataract formation—is a common complication of longstanding uveitis and/or chronic corticosteroid therapy.3
Finally, the vitreous may show cellular infiltration, "snowball opacities" (commonly seen in intermediate uveitis and sarcoidosis), fibrosis with resultant traction on the retina or cyclitic membrane formation behind the lens. It is important to determine the location of cells in the vitreous cavity. In cases of iridocyclitis, cells located in the anterior vitreous cavity simply may be “spillover,” whereas in cases of intermediate or posterior uveitis, cells are typically distributed either throughout the vitreous or more posteriorly. Posterior segment manifestations of uveitis may include disc and/or macular edema; retinal vasculitis; perivascular exudates; focal or diffuse retinitis or choroiditis; pars plana exudates (snowbanking); serous, tractional or rhegmatogenous retinal detachment; retinochoroidal atrophy; or choroidal and retinal neovascularization.1,3
Intraocular pressure can be affected by uveitis in many ways. Frequently, patients with acute iridocyclitis have low IOP due to inflammation-induced paralysis of the ciliary body and an associated decrease in aqueous production. Chronic uveitis or induced ciliary body detachment may result in hypotony and eventual phthisis bulbi. Additionally, IOP may be elevated secondary to posterior synechiae which can form between the pupillary rough and the lens creating a secluded pupil and when peripheral anterior synechiae form between the iris and angle blocking the aqueous from reaching the trabecular meshwork. Gonioscopy should be performed regularly in patients with chronic uveitis to check for peripheral anterior synechiae formation and secondary angle closure.3
Interestingly, 2% to 9% patients who have either ulcerative colitis or Crohn's disease develop uveitis, episcleritis or scleritis.1,2 Uveitis in association with inflammatory bowel disease (IBD) is frequently bilateral, often prolonged, may start insidiously, and usually has a posterior component. Uveitis in association with IBD is more common in women than in men. Nearly 25% of patients who have had uveitis in association with IBD fail to follow the sudden onset, recurrent, anterior, unilateral pattern that is characteristic of most HLA-B27-associated iritis. Overall, uveitis is far more likely to be associated with Crohn's disease than ulcerative colitis.1,4 Because uveitis may be a clue to the diagnosis of IBD, patients who have an iritis of unknown etiology should be questioned carefully regarding their bowel habits.1,4
We treated our patient with topical steroids q.i.d. and topical cyclopentolate 1% q.d. as well as a regimen of oral prednisone, which was comanaged by the medicine team. Stronger cycloplegics were not deemed necessary. The uveitis responded well and all medicines were discontinued over a three-week period.
Thanks to Spenser J. Brittain, O.D., of Monroe, Mich. for contributing this case.
1. Rosenbaum JT. Anterior Uveitis and Systemic Disease. In: Yanoff M, Duker JS (eds). Ophthalmology. Philadelphia: Mosby; 1999:1-8.
2. Cowen CL. Sarcoidosis. In: Yanoff M, Duker JS (eds). Ophthalmology. Philadelphia: Mosby; 1999:1-6.
3. Forster DJ. General Approach to The Uveitis Patient and Treatment Strategies. In: Yanoff M, Duker JS (eds). Ophthalmology. Philadelphia: Mosby; 1999:1-6.
4. Crawford JM. The Gastrointestinal Tract. In: Robbins SL, Cotran RS, Kumar V (eds). Pocket Companion to Robbin’s Pathologic Basis of Disease, 5th ed. Philadelphia, W.B. Saunders; 1995:311-41.







